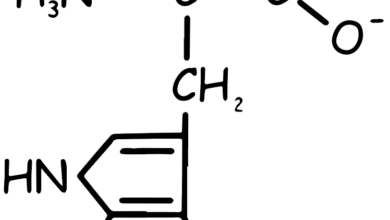
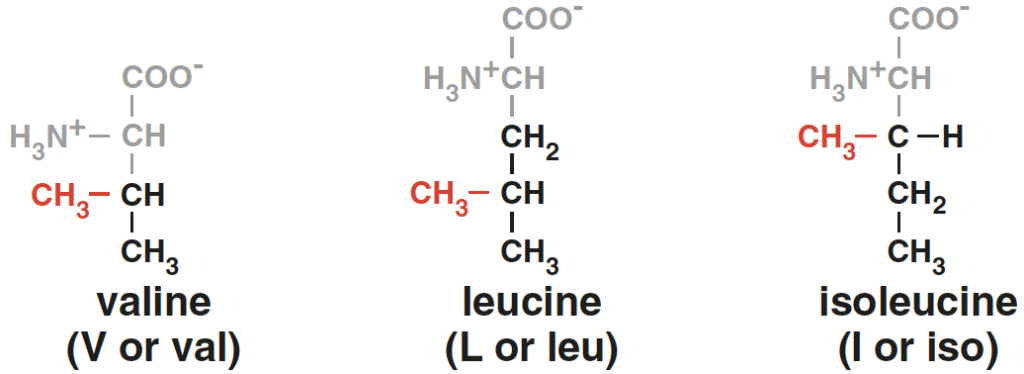
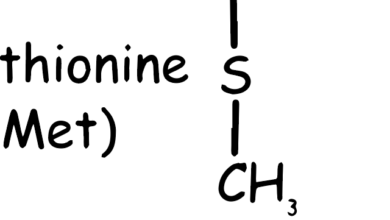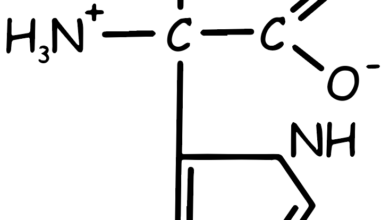Valine, Leucine and Isoleucine
These amino acids are termed “branched-chain” due to their chemical structure, which includes a branch off the main trunk of the molecule. Branched-chain amino acids (BCAAs) are a group of three essential amino acids, meaning they can’t be produced by your body and must be obtained from food.
BCAAs are found in protein-rich foods such as meat, dairy products, and legumes. They are also popular dietary supplements, especially among bodybuilders and athletes. However, most people can get enough BCAAs through their diet without needing supplements. As with any supplement, it’s important to consult a healthcare provider before starting BCAAs, especially for individuals with certain health conditions or those taking other medications.
Metazoans are incapable of producing BCAAs, yet these amino acids are prevalent in animals, making up about 35% of essential amino acids in most mammals. The branched, small, and hydrophobic nature of the R groups in BCAAs makes them essential in protein composition. Collectively, BCAAs constitute around 18% of amino acids and 63% of hydrophobic amino acids in proteins across various organisms.
Maple Syrup Urine Disease (MSUD)
Maple Syrup Urine Disease (MSUD) is a rare genetic disorder (occurrence 1 in 185,000) characterized by a deficiency in the body’s ability to break down certain amino acids: leucine, isoleucine, and valine. t involves either a partial or total deficiency in the enzyme complex known as branched-chain α-keto acid dehydrogenase. In MSUD, these amino acids and their associated α-keto acids build up in the blood, leading to toxicity that adversely affects brain functions. Symptoms include feeding difficulties, vomiting, dehydration, severe metabolic acidosis, and the urine’s distinctive maple syrup smell. Without treatment, MSUD can cause mental retardation, physical disabilities, and potentially death.

- Types of MSUD: The term encompasses both a classic form and various other variants. The classic type, which is the most prevalent, shows minimal to no activity of branched-chain α-keto acid dehydrogenase in leukocytes or cultured skin fibroblasts. Infants with the classic form exhibit symptoms in the initial days of life and face a high risk of fatality in early infancy without diagnosis and treatment. Intermediate forms, with 3–15% of normal enzyme activity, have less severe symptoms that can appear anytime from infancy to adulthood. A rare thiamine-dependent variant shows improved enzyme activity with high doses of thiamine.
- Screening and Diagnosis: Similar to PKU, prenatal and neonatal screenings are available, with most individuals being compound heterozygotes.
- Treatment: MSUD is managed with a specialized synthetic formula that provides limited but adequate amounts of leucine, isoleucine, and valine for normal growth and development, without reaching toxic levels. Prompt diagnosis and lifelong dietary management are crucial for normal development in children with MSUD. Notably, branched-chain amino acids are important energy sources during metabolic stress, and individuals with MSUD are vulnerable to decompensation during increased protein breakdown.
Isovaleric Acidemia (IVA)
Isovaleric Acidemia (IVA) is a genetic disorder caused by the deficiency of the enzyme isovaleryl-CoA dehydrogenase (IVD), which is crucial in the leucine degradation pathway. This deficiency leads to the accumulation of isovaleric acid and its derivatives, causing various metabolic issues. The primary treatment for IVA includes dietary restrictions (especially low protein and leucine diets), as well as supplementation with L-carnitine and glycine. Recent research has focused on genetic and biochemical approaches for prenatal diagnosis, understanding the molecular basis of the disease, and exploring new treatment strategies.
Overall, isovaleric acidemia, caused by a deficiency of IVD, can be effectively managed with early diagnosis and appropriate treatment strategies, leading to favorable outcomes in patients.