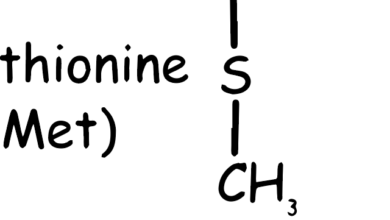Cysteine amino acid

Cysteine is a non-essential amino acid, which means that it can be synthesized by the human body and is not required to be obtained from the diet. It is notable for containing a sulfur atom, which is rare among amino acids. This sulfur allows cysteine to form disulfide bonds, which are important for the structure and function of proteins. Although cysteine can be synthesized in the body from methionine (another amino acid) via the transsulfuration pathway, it is also found in high-protein foods such as poultry, eggs, dairy, red peppers, garlic, onions, brussels sprouts, oats, and wheat germ. The balance of cysteine and other amino acids is important for various physiological processes and overall health.
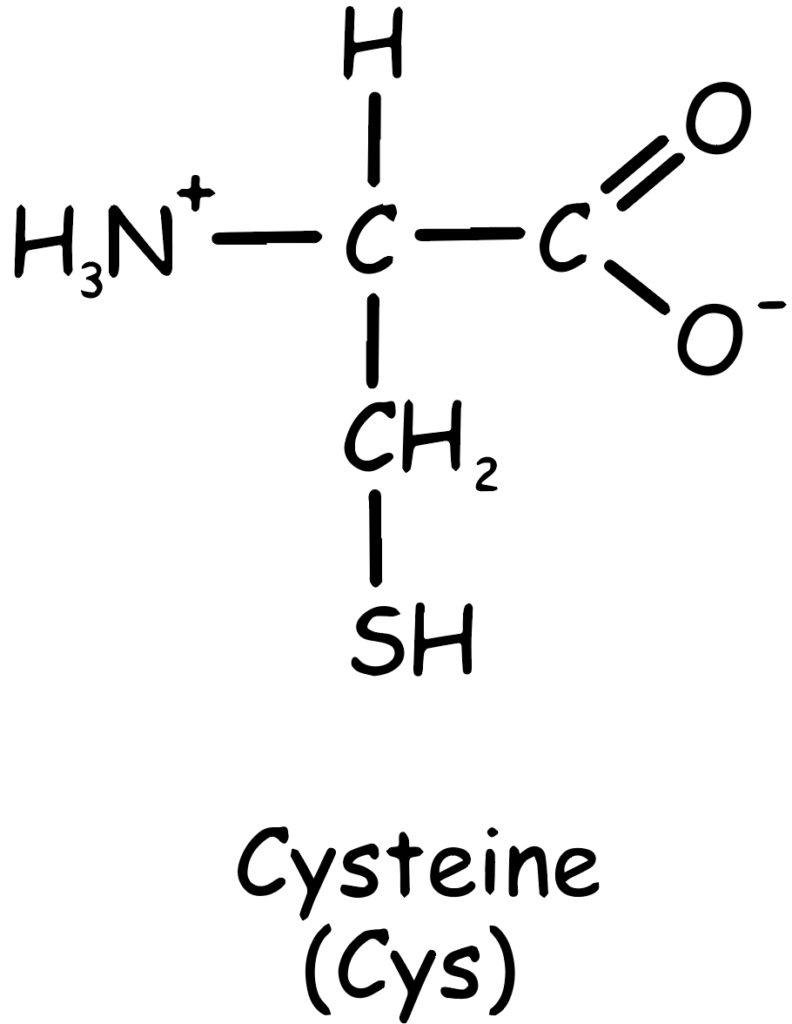
As shown in the Figure 1, the chemical structure of cysteine includes a carboxyl group (–COOH), an amino group (–NH2), and a side chain with a thiol group (–SH), making it highly reactive (Sulfhydryl group comes from methionine, C skeleton comes from methionine amino acid). This reactivity is crucial for the biological functions of cysteine.
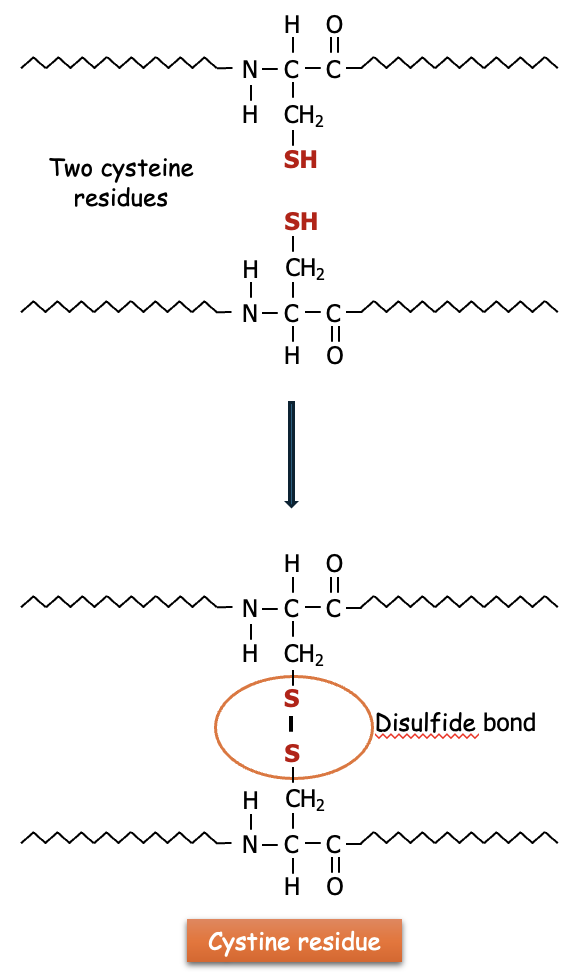
Protein Structure: The disulfide bonds formed between cysteine residues in proteins help stabilize the tertiary and quaternary structures of proteins. These bonds are crucial in the folding and stability of many proteins, including enzymes and antibodies.
Antioxidant Properties: Cysteine is a component of glutathione, a potent antioxidant that protects cells from oxidative stress. Glutathione helps neutralize free radicals and reactive oxygen species, thus playing a key role in maintaining cellular health.
Metal Ion Binding: Cysteine can bind to various metal ions due to its thiol group, playing a role in the transport and bioavailability of metals like iron and zinc.
Detoxification: Cysteine contributes to detoxification processes in the liver, as it is a component of various detoxifying enzymes.
Cystine
Cystine is created when two cysteine molecules become oxidized and form a covalent bond between their sulfur atoms. This disulfide bond (—S—S—) is what links the two cysteines together (see Figure 3). The disulfide bonds found in cystine are critical for maintaining the stability and structure of many proteins. Cystine is found in many proteins, particularly those secreted from cells or located in harsh environments (like digestive enzymes or hair keratin), where the disulfide bonds provide extra stability and resistance to denaturing conditions.
In certain conditions, cystine can accumulate and form crystals or stones in the kidneys or urinary tract, known as cystinuria. This is a genetic disorder that affects the reabsorption of cystine in the kidneys. Since cystine is formed from cysteine, foods rich in cysteine like meat, eggs, dairy products, nuts, seeds, and legumes can contribute to cystine levels in the body. Cystinuria is diagnosed through urine tests that show elevated levels of cystine and other dibasic amino acids (lysine, arginine, and ornithine). Genetic testing can also be used to confirm the diagnosis.
Management focuses on keeping the urine diluted and maintaining a high urinary output to prevent stone formation, usually through high fluid intake. Alkalization of urine and dietary modifications can also help. In some cases, medications that bind cystine in the urine are used. Surgical or endoscopic procedures may be required to remove large stones.
Cystinuria is a lifelong condition that requires ongoing management to prevent complications, particularly the formation of cystine stones, and to maintain kidney health.
Homocystinuria
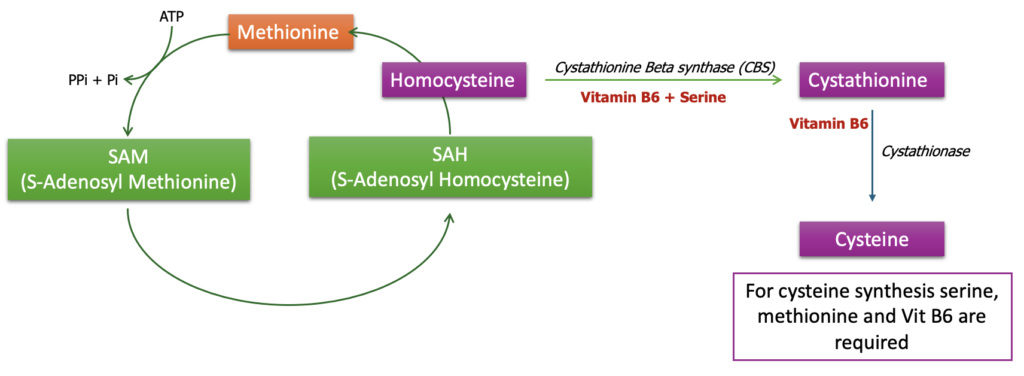
Homocystinuria is a rare genetic disorder characterized by an abnormal accumulation of the amino acid homocysteine in the blood and urine. This condition arises due to defects in the enzymes that are involved in the metabolism of methionine, particularly those involved in converting homocysteine into other substances. The most common form of homocystinuria is caused by a deficiency in the enzyme cystathionine β-synthase (CBS).
Homocystinuria is typically inherited in an autosomal recessive pattern, meaning that a child must inherit two copies of the mutated gene (one from each parent) to develop the condition. The symptoms of homocystinuria can vary but often include vision problems, skeletal abnormalities (such as long limbs and scoliosis), intellectual disability, and a predisposition to blood clots (thrombosis). The severity of symptoms can vary among individuals.
Homocystinuria is often diagnosed through newborn screening tests, which detect elevated levels of methionine or homocysteine. Genetic testing can confirm the diagnosis by identifying mutations in the relevant genes. reatment typically involves dietary modifications to limit methionine intake, supplementation with vitamins (such as B6, B12, and folic acid) that help reduce homocysteine levels, and, in some cases, medication to prevent blood clots. The specific treatment depends on the type of enzyme deficiency and the individual’s response to therapy.
With early diagnosis and appropriate treatment, many individuals with homocystinuria can lead relatively normal lives. However, untreated homocystinuria can lead to serious complications, including premature cardiovascular disease.
Cystinosis
Cystinosis is a rare, inherited metabolic disorder characterized by the abnormal accumulation of the amino acid cystine within cells. This buildup leads to the formation of cystine crystals, which can cause damage to various organs and tissues.
Cystinosis is caused by mutations in the CTNS gene, which encodes for a protein called cystinosin. This protein is responsible for transporting cystine out of lysosomes, the cellular compartments that break down waste materials. When cystinosin is dysfunctional or absent, cystine accumulates in the lysosomes.
Common symptoms include kidney problems, growth retardation, rickets (due to renal phosphate loss), and photophobia due to cystine crystals in the eyes. If left untreated, nephropathic cystinosis can lead to kidney failure and other complications.
Regular monitoring and treatment can improve quality of life and life expectancy. Early and consistent treatment is especially crucial in managing the nephropathic form of cystinosis. Cystinosis is a lifelong condition that requires ongoing management and care. Advances in treatment have significantly improved outcomes for individuals with this disorder.