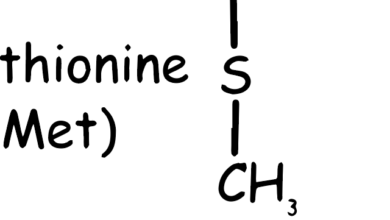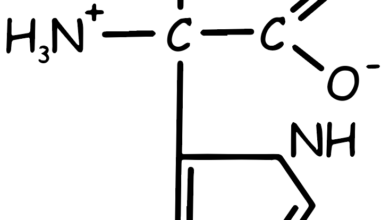Tyrosine Amino Acid
Tyrosine is an amino acid that plays a crucial role in protein synthesis. Its molecular structure consists of a phenolic hydroxyl group (-OH) attached to a benzene ring, which in turn is linked to the α-carbon of the amino acid (See Figure 1). This structure gives tyrosine its characteristic properties and enables it to participate in various biochemical processes.
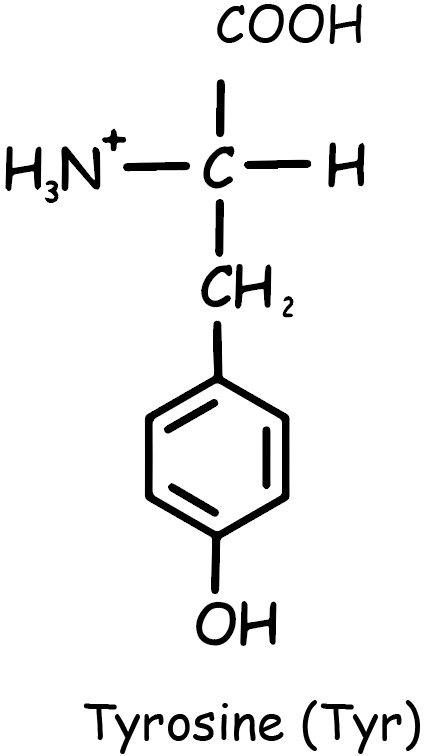
Chemically, tyrosine is designated as 4-hydroxyphenylalanine, indicating its relationship to phenylalanine with the addition of a hydroxyl group. This modification significantly influences its role in biological systems. Tyrosine is involved in the synthesis of important neurotransmitters like dopamine and norepinephrine, and hormones such as thyroid hormones. It is also a precursor for the pigment melanin.
In terms of its general properties, tyrosine is a non-essential amino acid, meaning the body can synthesize it as needed, usually from phenylalanine. It is polar due to the hydroxyl group, but the aromatic ring also gives it a degree of hydrophobicity, making it a versatile component in protein structures.
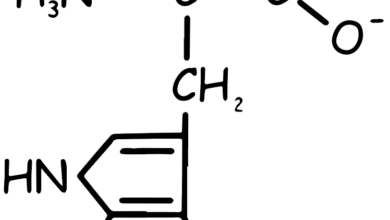
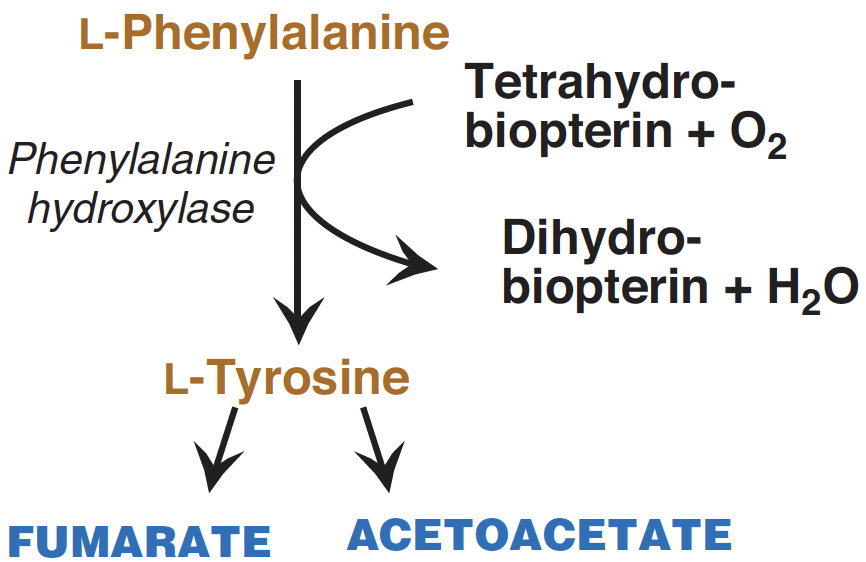
The conversion of phenylalanine into tyrosine (see Figure 2) is achieved through hydroxylation, a process driven by the enzyme phenylalanine hydroxylase, which requires tetrahydrobiopterin. This reaction marks the beginning of phenylalanine breakdown. Consequently, the metabolic pathways of phenylalanine and tyrosine converge, resulting in the production of fumarate and acetoacetate. As a result, both phenylalanine and tyrosine have the dual capacity to be glucogenic and ketogenic.
Phenylketonuria
Phenylketonuria (PKU), also known as phenylalanine hydroxylase (PAH) deficiency, is an inherited autosomal recessive disorder affecting phenylalanine metabolism. Inherited deficiencies in the enzymes of phenylalanine and tyrosine metabolism lead to the disease (See Figure 3). High levels of phenylalanine can lead to brain dysfunction, causing severe intellectual disability, epilepsy, and behavioral issues if not treated. The disorder occurs in approximately 1 out of every 10,000 newborns globally.
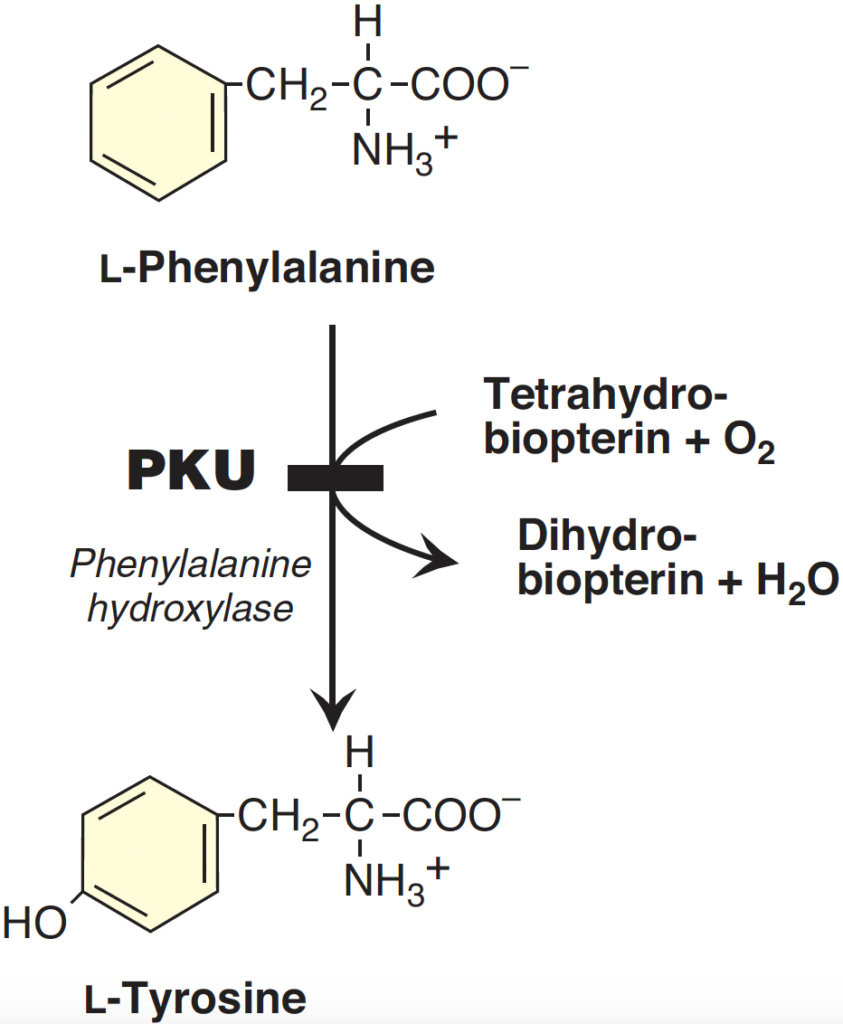
Early detection through newborn screening and prompt treatment initiation can maintain normal intelligence levels, although some neurocognitive challenges may persist.
For over six decades, dietary phenylalanine restriction has been the primary treatment, proving effective but challenging for patients to consistently follow. Alternative treatments include tetrahydrobiopterin, beneficial for a small group of patients, typically those with less severe PKU, and pegylated phenylalanine ammonia lyase, which requires daily injections and can trigger adverse immune reactions. Ongoing research explores new treatments like mRNA and gene therapy, addressing the limitations of current methods. Despite being the most common amino acid metabolism disorder, the brain dysfunction in PKU patients remains poorly understood, highlighting the need for further research to develop treatments based on a deeper understanding of the disease’s pathophysiology.
The recent advancement of tandem mass spectrometry in neonatal screening has significantly aided in the early detection and treatment of Phenylketonuria (PKU), which helps in reducing the risk of mental retardation commonly associated with this condition.
Diagnosis of PKU
The FeCl3 test, which is less sensitive, is used to detect Phenyl Pyruvate in urine (a keto acid), and a positive result yields a blue-green color.
In comparison, Guthrie’s test, also known as the Bacterial inhibition test, is more accurate than the FeCl3 urine test. This test utilizes Bacillus subtilis to detect serum phenylalanine levels. However, for reliable results, Guthrie’s test should be conducted 1-2 weeks post-birth, after the infant has consumed a diet containing the amino acid phenylalanine (found in both human and cow’s milk).
For a definitive diagnosis, the measurement of amino acid levels in the blood through Tandem Mass Spectrometry is employed. This method can detect even minor increases in phenylalanine present at birth.
In phenylketonuria (PKU), the brain is affected due to:
- The excessive accumulation of phenylalanine in the blood, which then enters the brain via the L-aromatic amino acid transporter in the brain’s capillaries. This leads to a competitive inhibition, preventing other aromatic amino acids like Tyrosine and Tryptophan from entering the brain. These amino acids are crucial for synthesizing various neurotransmitters needed in the brain.
- A shortage of Thyroxine, which is derived from Tyrosine.
- A decrease in cerebral serotonin levels, since serotonin is produced from Tryptophan.
Catecholamines and serotonin are important neurotransmitters in the body, and their synthesis involves the amino acid tyrosine.